
- Executive summary
- Introduction
- Overview of approach
- Value to patients
- Value to economic growth
- Value to the health and social care system
- The case for change
- Annex A Methodology: Description of scenarios
- Annex B Methodology: Value to patients
- Annex C Methodology: Value to economic growth
- Annex D Methodology: Valuing impact on Carer’s Allowance
- Annex E Results: Additional modelled scenarios
- References
- Executive summary
- Introduction
- Overview of approach
- Value to patients
- Value to economic growth
- Value to the health and social care system
- The case for change
- Annex A Methodology: Description of scenarios
- Annex B Methodology: Value to patients
- Annex C Methodology: Value to economic growth
- Annex D Methodology: Valuing impact on Carer’s Allowance
- Annex E Results: Additional modelled scenarios
- References
Executive summary
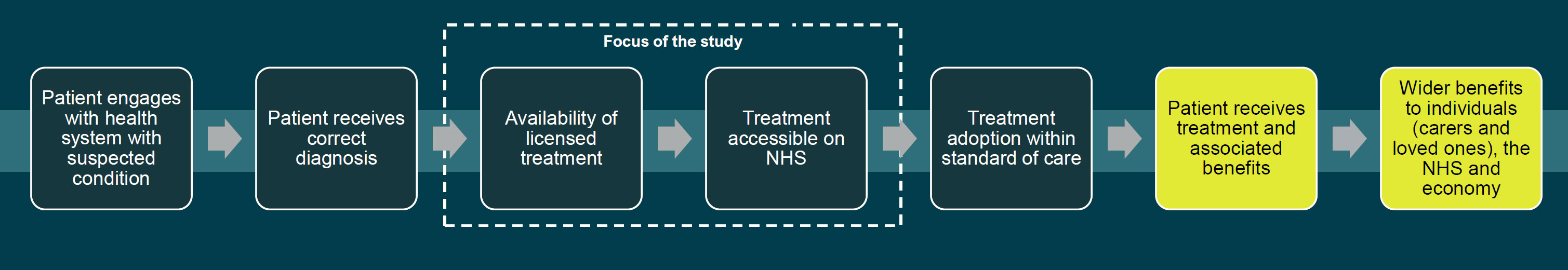


1 Introduction
- The awareness challenge. There are between 6,000 and 8,000 individual rare diseases. This means that, while individually rare, together there are over 3.5 million people in the UK living with a rare disease.13 Raising awareness of rare diseases is a key step in improving access to care and resources.
- The diagnosis challenge. On average people have to wait 5.6 years after onset of symptoms to receive a rare disease diagnosis.14 This delays the ability of people to access care and support for their condition, and the diagnosis process itself can often have a significant impact on a person's quality of life.
- The evaluation and access to medicines challenge. Only 5 per cent of rare diseases have an effective treatment.15 Even where these do exist, fewer than 35 per cent of treatments licensed in the EU are fully accessible (inclusion on the public reimbursement list) to patients in England and Scotland, and similar levels for NI and Wales who typically adopt NICE recommendations.16 Taking into account medicines licensed elsewhere but not in the EU, this percentage for full access in England and Scotland would be even lower; for example, evidence on FDA and EMA orphan medicine approvals between 2000 and 2022 found that just under 30 per cent of orphan medicines approved by the FDA were either not listed by the EMA or refused EMA authorisation.17 18 This means that there is significant unmet need amongst rare disease patients even after receiving a diagnosis.
- Helping patients get a final diagnosis faster;
- Increasing awareness of rare disease amongst healthcare professionals;
- Better coordination of care; and
- Improving access to specialist care, treatments and drugs.
2 Overview of approach
- Base case: This reflects the current clinical and policy environment.
- Enhanced access scenario: This scenario reflects two advances relative to the base case: (1) an expansion in the availability of licensed treatment, informed by the current orphan medicines pipeline, and (2) improved treatment access via NHS reimbursement, modelled as the UK reaching the top three European countries for access to licensed orphan medicines, in line with UK Life Sciences Sector Plan ambitions to be amongst the top three countries in Europe for speed of patient access to medicines.
- Best in Europe access scenario: Compared to the enhanced scenario, this scenario models the impact of the UK becoming the best country in Europe for access to orphan medicines on the NHS.
- Spinal muscular atrophy (SMA)
- Neuromyelitis optica spectrum disorder (NMOSD)
- Motor neurone disease (MND)
| Scenario | % of patients for whom a licensed treatment is available | per cent of treatments fully accessible on the NHS |
|---|---|---|
| Base | 5% | 35% |
| Enhanced access | 13% | 50% |
| Best in Europe access | 13% | 89% |
- Treatment availability. Currently only around 5 per cent of rare diseases have an effective treatment.23
- Treatment access in the UK. Research has found that of those orphan medicines licensed by the European Medicines Agency (EMA), only 38 per cent and 32 per cent are fully available (inclusion on the public reimbursement list) to patients on the NHS in England and Scotland respectively.24 Given NICE guidance is reflected in Wales and Northern Ireland, we take the average of these as a proxy of average orphan medicine reimbursement rates in the UK.
- Treatment availability. Increase in the proportion of patients who have a treatable rare disease from 5 per cent to 13 per cent based on evidence on the orphan medicine pipeline. An analysis of US Food and Drug Administration (FDA) data found that up to 11–15 per cent of rare diseases have at least one medicine that has been developed and shown promise in their treatment, diagnosis or prevention.25 We take the average of this range to model the potential increase in treatment availability.
- Treatment access in the UK. The UK Life Sciences Sector Plan sets out an ambition that, by 2030, the UK will be one of the top three fastest places in Europe for patient access to medicines and MedTech. Looking across 2020–2023, the third highest full availability rate for orphan medicines in Europe (measured as a per cent of all EMA licensed products) was 50 per cent, which forms the basis of this scenario.
- Treatment availability. Same assumptions as in the enhanced access scenario.
- Treatment access in the UK. Across 2020–2023, Germany had the highest full availability rate for orphan medicines at 89 per cent, which forms the basis of this scenario.
3 Value to patients
- Under the assumption that the UK moves to the top three best countries for treatment access, improving treatment access via NHS reimbursement alone leads to additional QALY benefits of £0.4 billion.
- Assuming, instead, that the UK becomes the best in Europe for treatment access, additional lifetime QALY benefits amount to £1.3 billion.


- Assumptions on the diagnostic odyssey. The analysis is based on the current experience of rare patients in receiving a final diagnosis. However, diagnosis is a major challenge for accessing treatment, with the average patient waiting 5.6 years from the onset of symptoms before receiving a diagnosis.32 The NHS 10 Year Plan sets out an ambitious commitment to improving the diagnosis of rare disease.33 If achieved, this would increase the number of patients able to access treatments, thereby amplifying the benefits estimated under the same modelling assumptions.
- Available data on prevalence. Diagnosed prevalence of rare diseases in the UK is estimated at 1.5 per cent, compared with a global point prevalence of approximately 3.5 per cent reported in Orphanet.34 This gap is partly driven by the diagnostic odyssey described above and partly by limitations in NHS electronic health record coverage and clinical coding, which constrain the identification of a large share of rare diseases. As a result, prevalence estimates based on diagnosed patients are likely to understate the true population affected.
- QALY benefit of new treatments. The QALY gains associated with treatment discovery are based on backward-looking evidence from orphan medicine approvals between 1999 and 2019. Over this period, the median incremental lifetime QALY gain reported for orphan medicines is 0.43, which is the value used in this analysis. However, if we were to take the mean QALY estimate rather than the median from this same review, this would give us an incremental lifetime QALY gain of 0.99 (further details in Annex E). The mean QALY estimate is higher than the median as it reflects the small number of breakthrough treatments to date that can generate significant QALY gains. As we see more developments in areas such as gene therapy and other advanced therapies that could deliver potentially curative treatments for rare diseases, it is possible that future innovations will deliver larger average health gains than those observed historically so the mean QALY estimate may be a reasonable forward-looking perspective.
4 Value to economic growth



- Under the Enhanced access scenario, with access reimbursement rates in the top three in Europe, annual productivity benefits increase to approximately £1.1 billion annually (discounted).
- Moving to the more ambitious, Best in Europe access scenario, would deliver £2.3 billion of productivity benefits (discounted).
- Under this scenario, improving access through reimbursement alone, with treatment availability unchanged, would generate £0.8 billion a year in additional GVA, driven by improved labour market participation among working-age patients and carers of children with a rare disease.


5 Value to the health and social care system
- Impact on overall costs. In the UK, NICE's HST24 committee papers show that a patient in the "broad range of normal development (BRND)" health state is modelled to use far fewer health and care resources than someone in more severe SMA states. In the economic model, annual costs are around £8,333 in BRND, compared with about £283,710 for the "non-sitter + permanent assisted ventilation (PAV)" state.62
- Impact on hospital admissions and emergency visits. A study on the impact of recent DMTs found that inpatient admissions reduced by 41 per cent for children and 67 per cent for adults in the 12 months post-treatment compared to the 12 months pre-treatment, and emergency department visits decreased by 8 per cent.63
- Impact on hospitalisation rate and lengths of stays. Research on the impact of recent DMTs found that patients who received the treatment spent less time hospitalised (2 per cent versus 14 per cent) and the overall hospitalisation rate was half that of untreated patients. Patients receiving treatment had a lower average length of stay compared to untreated patients (7 versus 13 days).64
6 The case for change



Annex A Methodology: Description of scenarios
- Treatment availability: whether a licensed treatment exists for a given rare disease; and
- Treatment access: whether licensed treatments are fully reimbursed and routinely available to patients through the NHS.
- Base case: This reflects the current clinical and policy environment.
- Enhanced access scenario: This scenario reflects two advances relative to the base case: (1) an expansion in the number of rare diseases with a licensed treatment, informed by the current orphan medicines pipeline, and (2) the UK achieving its Life Sciences Sector Plan ambitions to be amongst the top three countries in Europe for speed of patient access to medicines.
- Best in Europe access scenario: Compared to the enhanced scenario, this scenario models the impact of the UK becoming the best country in Europe for accessibility of orphan medicines on the NHS.
- Base case: 35 per cent based on an average of England and Scotland full availability rate for orphan medicines, and assuming the figure for England is reflected in Wales and Northern Ireland;
- Enhanced access scenario: Top three in Europe (benchmarking to the third highest-performing country, Austria, 50 per cent full availability); and
- Best in Europe access scenario: Best in Europe (benchmarking to the highest-performing country, Germany, 89 per cent).
- Treatment availability (the proportion of rare diseases with a licensed treatment); and/or
- Treatment access (the proportion of licensed treatments that are fully available on the NHS).
Annex B Methodology: Value to patients
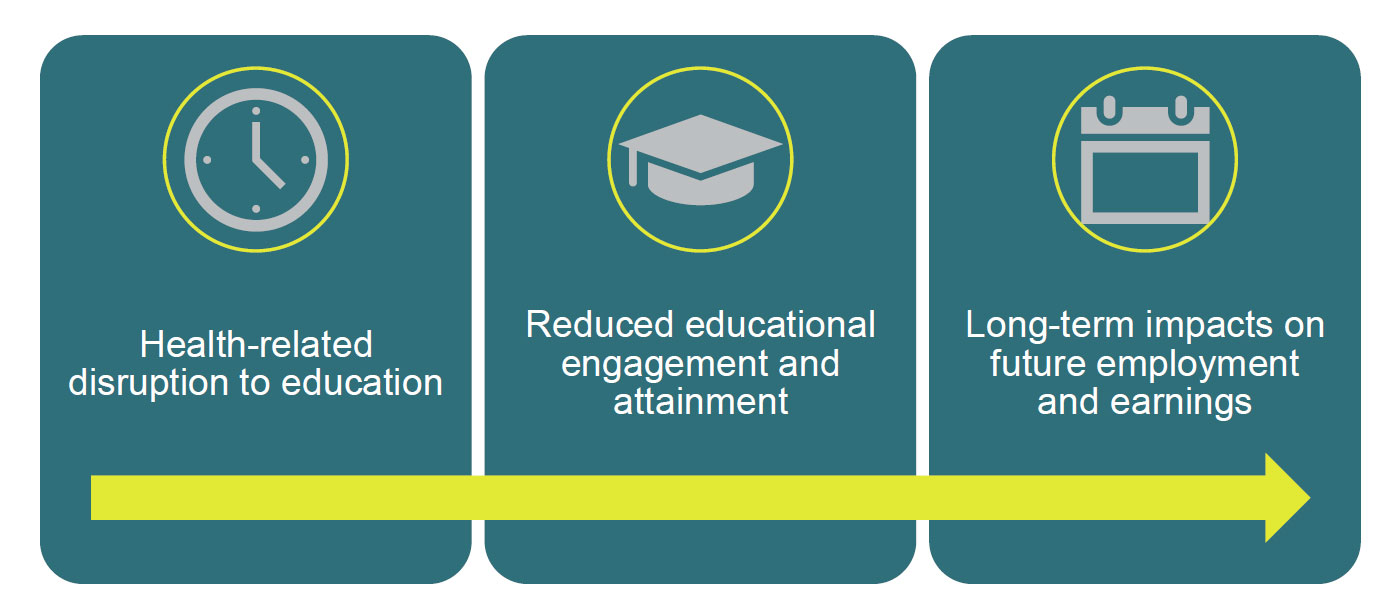
- Estimate the number of patients receiving treatment under each scenario and base case;
- Apply evidence-based incremental QALY gains per treated patient;
- Value incremental QALYs using Treasury Green Book QALY values; and
- Apply discounting to reflect the timing of treatment availability for benefits associated with treatment discovery.
- The size of the rare disease population;
- The proportion of patients diagnosed;
- The proportion of rare diseases for which a treatment exists (treatment availability); and
- The proportion of eligible patients who are able to access treatment via NHS reimbursement (treatment access).
| Outcomes | Orphan medicines | Ultraorphan medicines | Total |
|---|---|---|---|
| Number of medicine-indication pairs | 73 | 33 | 106 |
| Incremental QALYs: Median (IQR) | 0.31 (0.76) | 0.70 (1.43) | 0.43 |
| Incremental QALYs: Mean (SD) | 0.58 (1.13) | 1.89 (5.13) | 0.99 |
- Central estimate: £70,000 per QALY (2020/21 prices), based on Green Book supplementary wellbeing guidance. This value reflects a cross-government perspective on the societal value of health improvements.
- Lower bound: £30,000 per QALY, reflecting the midpoint of NICE standard thresholds for non-rare conditions, as per the update planned for April 2026.
- Upper bound: £85,000 per QALY, representing an inflation-adjusted Green Book value (2025/26 prices) to reflect more recent price levels.
- Health benefits associated with treatments that are not yet available to patients are discounted to reflect the time lag between treatment development and realisation of health gains. This is consistent with HM Treasury Green Book guidance, which recommends discounting future health and life outcomes at 1.5 per cent per annum.
- For treatments that are already available, patient health benefits are assumed to accrue immediately and are therefore not discounted. However, for pipeline orphan medicines, health benefits will only be realised once treatments progress through regulatory approval and become accessible to patients in routine care. Discounting is applied to reflect this delay.
- The average clinical development duration is 7.2 years.
- Following approval, there is an additional delay before routine patient access. Based on UK evidence on patient access timelines, the average time from approval to access is assumed to be 1.07 years.
| Item | Base case | Enhanced access | Best in Europe access | Source |
|---|---|---|---|---|
| UK population (A) | 69,300,000 | ONS | ||
| Per cent diagnosed with rare disease (B) | 1.50% | Thygesen et al, 2025 | ||
| UK patients diagnosed with rare disease (C) | 1,039,500 | 1,039,500 | 1,039,500 | C = A*B |
| Rare cancer uplift (D) | 55% | Rare condition statistics, NHS | ||
| UK patients diagnosed with rare disease (incl. rare cancer) (E) | 1,611,225 | 1,611,225 | 1,611,225 | E = C*(1+D) |
| Treatment exists to manage condition (F) | 5% | 13% | 13% | UK govt. (base), Fermaglich and Miller, 2023 (scenario) |
| Patient access (% public availability) (G) | 35% | 50% | 89% | Patient W.A.I.T Indicators (base and scenario) |
| Patients with diagnosis and treatment (H) | 28,196 | 104,730 | 186,419 | H = E*F*G |
| QALY benefit with treatment (I) | 0.43 | 0.43 | 0.43 | Clifford et al, 2024 |
| Total QALYs (J) | 12,124 | 45,034 | 80,160 | J = H*I |
| Value per QALY: Green Book appraisal value (in £), unadjusted (K) | £70,000 (£30,000 – £87,500) | |||
| HRQoL value (£billion) (L) | £0.9 (£0.4 – £1.1) | £3.2 (£1.4 – £3.9) | £5.6 (£2.4 – £7.0) | L = J*K |
| Delta HRQoL value (£billion) (M) | £2.3 (£1.0 – £2.9) | £4.8 (£2.0 – £6.0) | M = Lscenario – Lbase | |
| Delta HRQoL value (£billion, discounted) (N) | £2.1 (£0.9 – £2.6) | £4.4 (£1.9 – £5.5) | Green Book discount rate for health and life outcomes | |
Annex C Methodology: Value to economic growth
- Estimate productivity loss associated with unmanaged rare disease, for patients and carers;
- Apply evidence-based reductions in productivity loss associated with effective treatment;
- Scale productivity gains by the number of additional patients gaining access to treatment under each scenario; and
- Apply discounting to reflect the timing of benefit realisation and estimate cumulative five- and ten-year productivity benefits.
- Total annual GVA lost due to reduced workforce participation and absenteeism; and
- Average productivity loss per unmanaged patient or per carer.
- The diagnosed rare disease population;
- The proportion of rare diseases for which a treatment exists (treatment discovery) in the base case and in each scenario; and
- The proportion of eligible patients able to access treatment (treatment access) in the base case and in each scenario.
- Productivity benefits associated with improved access to existing treatments are assumed to accrue immediately.
- Productivity benefits associated with new treatment discovery are discounted to reflect the time lag between orphan designation, regulatory approval and routine patient access.
| Source | Working-age patients | Carers | Total (£billion) | Source | |
|---|---|---|---|---|---|
| Productivity loss per unmanaged patient | (A) | £48,025 | £36,308 | Survey evidence | |
| % remaining productivity loss | (B) | 51% | 20% | Chiesi, 2022; Chiesi, 2023 † | |
| Productivity loss per managed patient | (C) | £24,406 | £7,260 | (C) = (A)*(B) | |
| Reduction in productivity loss per patient | (D) | £23,619 | £29,047 | (D) = (A) – (C) | |
| Enhanced access scenario | (F) | £0.8 | £0.6 | £1.1 (discounted) | (D)*(Δ treated patients) |
| Best in Europe access scenario | (G) | £1.6 | £1.2 | £2.3 (discounted) | (D)*(Δ treated patients) |
Annex D Methodology: Valuing impact on Carer's Allowance
- The number of children diagnosed with rare disease;
- The proportion without access to treatments;
- Evidence on labour market participation among parents of children with rare disease.
Annex E Results: Additional modelled scenarios
- Enhanced access. The increase in treatment access from better reimbursement rates on the NHS combined with the expected expansion in licensed treatment availability leads to £2.1 billion of additional QALY benefits compared to the base case, and to additional annual productivity benefits of £1.1 billion (discounted).
- Best in Europe access. Under the pipeline-informed treatment availability assumption, combined with best in Europe access reimbursement rates, additional QALY benefits amount to £4.4 billion, while annual productivity benefits increase to approximately £2.3 billion annually (discounted).
- Additional scenario: using mean rather than median lifetime QALY gain. We model an additional scenario where we assume the mean rather than the median lifetime QALY gain (0.99 vs. 0.43 QALYs per treated patient). Our estimates indicate that this would increase lifetime benefits from £4.4 billion to £10.0 billion in the best in Europe access scenario. The mean QALY estimate is higher than the median as it reflects the small number of breakthrough treatments that can generate significant QALY gains. Both the mean and median QALY estimates are taken from the same review, based on the distribution of benefits observed across treatment approvals from 1999 to 2019. The mean may be a reasonable forward-looking perspective if new rare disease treatments are more likely to resemble the higher-impact therapies seen in the historical sample.
- Additional scenario: improvement in reimbursement rates only (no improvement in treatment availability). We model an additional scenario in which reimbursement rates improved while treatment availability remains at current levels (i.e. 5 per cent of rare diseases have a licensed treatment without assuming expansion from the pipeline). Under the assumption that the UK moves to the top three in treatment access, improving treatment access via NHS reimbursement alone leads to additional QALY benefits of £0.4 billion. Assuming, instead, that the UK becomes the best in Europe, additional lifetime QALY benefits amount to £1.3 billion.
- Additional scenario: estimate of benefits to patients and to the economy excluding rare cancers. We have modelled an additional scenario where benefits to patients and to the economy are computed excluding rare cancers from the list of considered conditions. Table 5 and Table 6 report detailed results for estimated benefits to patients and productivity benefits of this scenario.
| Scenarios | Patient benefits (lifetime Health-Related Quality of Life value), (£billion, discounted) | Productivity benefits (annual), (£billion, discounted) | ||
|---|---|---|---|---|
| Enhanced access | Best in Europe access | Enhanced access | Best in Europe access | |
| Core scenario | £2.1 | £4.4 | £1.1 | £2.3 |
| Adopting mean QALY gain as opposed to median | £4.8 | £10.0 | £1.1 | £2.3 |
| Improvement in reimbursement rates only | £0.4 | £1.3 | £0.2 | £0.8 |
| Core scenario excluding rare cancer | £1.4 | £2.8 | £0.7 | £1.5 |
Note: Summary of scenario analysis, estimation steps detailed in more detail in Table 2 and Table 5
| Item | Base case | Enhanced access | Best in Europe access | Source | |
|---|---|---|---|---|---|
| UK population | (A) | 69,300,000 | ONS | ||
| % diagnosed with rare disease | (B) | 1.50% | Thygesen et al, 2025 | ||
| UK patients diagnosed with rare disease | (C) | 1,039,500 | 1,039,500 | 1,039,500 | C = A*B |
| Treatment exists to manage condition | (D) | 5% | 13% | 13% | UK govt. (base), Fermaglich and Miller, 2023 † (scenario) |
| Patient access (% public availability) | (E) | 35% | 50% | 89% | Patient W.A.I.T Indicators (base and scenario) |
| Patients with diagnosis and treatment | (F) | 18,191 | 67,568 | 120,270 | F = D*E*F |
| QALY benefit with treatment | (G) | 0.43 | 0.43 | 0.43 | Clifford et al, 2024 |
| Total QALYs | (H) | 7,822 | 29,054 | 51,716 | H = F*G |
| Value per QALY: Green Book appraisal value (in £), unadjusted (typical NICE values, Green Book value adjusted) | (I) | £70,000 (£30,000 – £87,500) | |||
| HRQoL value (£billion) | (J) | £0.6 (£0.2 – £0.7) |
£2.0 (£0.9 – £2.6) |
£3.6 (£1.6 – £4.5) |
J = H*I |
| Delta HRQoL value (£billion) | (K) | £1.5 (£0.6 – £1.9) |
£3.1 (£1.3 – £3.9) |
K = Lscenario – Lbase | |
| Delta HRQoL value (£billion, discounted) | (L) | £1.4 (£0.6 – £1.7) |
£2.8 (£1.2 – £3.5) |
Green Book discount rate for health and life outcomes | |
Note: Estimates shown according to central QALY value of £70,000, analysis was also run using range of £30,000 – £87,500. † Uses the US jurisdiction definition of rare disease (<200,000 affected in the US)
| Source | Working-age patients | Carers | Total (£billion) | Source | |
|---|---|---|---|---|---|
| Productivity loss per unmanaged patient | (A) | £48,025 | £36,308 | Survey evidence | |
| % remaining productivity loss | (B) | 51% | 20% | Chiesi, 2022; Chiesi, 2023 † | |
| Productivity loss per managed patient | (C) | £24,406 | £7,260 | (C) = (A)*(B) | |
| Reduction in productivity loss per patient | (D) | £23,619 | £29,047 | (D) = (A) – (C) | |
| Enhanced access scenario | (F) | £0.5 | £0.4 | £0.7 (discounted) | (D)*(Δ treated patients) |
| Best in Europe access scenario | (G) | £1.0 | £0.8 | £1.5 (discounted) | (D)*(Δ treated patients) |
Note: † Uses the US jurisdiction definition of rare disease (<200,000 affected in the US).
References
-
KeywordsRare diseases
-
PublisherABPI, Frontier Economics
-
Last modified01 April 2026
-
Last reviewed01 April 2026
Related content

News |
ABPI response to publication of England's Rare Diseases Action Plan 2026
The ABPI has responded to England's latest Rare Diseases Action Plan, published in advance of Rare Disease Day.
News |
ABPI welcomes rare diseases research landscape report
The National Institute for Health and Care Research (NIHR) has today published the first Rare Diseases Research Landscape report for the UK.

News |
ABPI response to England's 2023 Rare Disease Action Plan
The Department of Health and Social Care has published the 2023 England Rare Diseases Action Plan, setting out the actions that will be delivered this year to support people with rare diseases in England.